背景技术:
聚乙烯亚胺是一种水溶性接枝聚合物,它的分子链上拥有大量的胺基,最突出的特点是其高阳离子电荷密度。从结构上聚乙烯亚胺可分为线性聚乙烯亚胺(l-pei)和支化聚乙烯亚胺(b-pei),其中线性聚乙烯亚胺分子链上的胺全部为仲胺,而支化聚乙烯亚胺分子链上的胺分为伯胺、仲胺和叔胺。目前,聚乙烯亚胺在基因转染、造纸、絮凝剂、染料固色剂以及纤维改性等领域有着广泛的应用。近年来,由于聚乙烯亚胺出色的吸附性能,其作为新型金属离子配位吸附材料受到了广泛关注。然而,聚乙烯亚胺为水溶性粘稠液体,在水相中以游离分子态形式存在,作为吸附剂存在操作困难、不易分离回收、易流失等缺点。
聚乙烯亚胺的化学修饰改性能极大地提高聚乙烯亚胺的性能和应用范围。目前,聚乙烯亚胺接枝的方法是利用聚乙烯亚胺上的伯胺氨基和仲胺氨基对其进行后修饰,常见的方法包括与羧酸的偶联反应、亲核取代反应、开环反应和迈克尔加成反应等,以及与端基官能化的聚合物反应或接枝聚合反应。然而,改性的聚乙烯亚胺聚合物分离纯化操作都较为繁琐,一般需要使用透析、冷冻干燥等方法除掉过量的未反应的试剂、副产物、溶剂和水,不适宜产品的大规模生产与应用。
n-磺酰基氮丙啶(氮杂环丙烷)化合物是一类非常重要的三元杂环化合物,因为它是构建复杂的含氮化合物的基石,也是重要的官能团转化中心。n-磺酰基氮丙啶化合物的开环反应一般需要在碱性条件下亲核开环或者在过渡金属等路易斯酸催化开环。以磺酰基活化的仲胺作为引发剂,六甲基二硅氨基钾作为碱,n-磺酰基氮丙啶可以发生开环聚合反应合成窄分布的聚磺酰胺高分子。然而,这些n-磺酰基氮丙啶开环聚合反应需要在无水无氧的条件下进行,且需要使用价格昂贵的引发剂,制备条件相对苛刻。
技术实现要素:
有鉴于此,本申请提供了一种改性聚乙烯亚胺聚合物及其制备方法,制备条件温和且产率高,无需反应溶剂,无需分离纯化操作,能够得到用于金属离子吸附的改性聚乙烯亚胺聚合物。
本申请的具体技术方案如下:
本申请提供一种改性聚乙烯亚胺聚合物,具有式(ⅰ)所示结构;
其中,m、n为正整数,10≤m+n≤600,r1为开环后的氮丙啶化合物,r3为r1或h,a、b、c为正整数,0≤a≤2,0≤b≤2,0≤c≤2,且r3为h时,a、b、c不同时为0。
本申请中,改性聚乙烯亚胺聚合物为浅黄色非粘稠固体,相较于商业化的聚乙烯亚胺更容易实现后加工处理。可选择性制备双亲性聚合物或油溶性聚合物,其中,双亲性聚合物用于金属离子吸附的吸附效率高,且回收再利用时吸附效率基本不变,在吸附材料、分析检测等领域具有较大的应用前景;油溶性聚合物则表现出优异的荧光性能。
优选的,r1具有式(ⅱ)所示结构;
其中,x为大于0的整数,r为h、烷基或芳基,r2为对甲苯磺酰基、甲磺酰基或吡啶磺酰基;
当x为1时,所述改性聚乙烯亚胺聚合物为双亲性聚合物;当x大于1时,所述改性聚乙烯亚胺聚合物为油溶性聚合物。
本申请中,r2作为吸电子取代基,降低了氮丙啶上的电子云密度,使其容易与聚乙烯亚胺的伯胺和仲胺发生亲核开环反应,在不严格控制反应条件的同时也能够提高产物的收率。n-磺酰基氮丙啶的当量较低时,聚乙烯亚胺引发n-磺酰基氮丙啶发生亲核开环反应,式(ⅱ)中的x为1,得到双亲性聚合物。当提高n-磺酰基氮丙啶的当量,聚乙烯亚胺中的叔胺作为有机碱,伯胺和仲胺作为引发位点来引发n-磺酰基氮丙啶发生阴离子开环聚合,能够提高聚氮丙啶的链长度,式(ⅱ)中的x为大于1的整数,得到油溶性聚合物。
优选的,r为h、碳原子数为1~15的烷基、
本申请中,选择这些取代基可增加疏水链的分子量,进而利于改善聚乙烯亚胺聚合物的性能。
优选的,所述改性聚乙烯亚胺聚合物的重均分子量大于4500。
本申请还提供一种改性聚乙烯亚胺聚合物的制备方法,由n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合,熔融反应制得所述改性聚乙烯亚胺聚合物。
本申请中,利用支化聚乙烯亚胺的伯胺和仲胺位点引发n-磺酰基氮丙啶化合物开环合成改性聚乙烯亚胺,由于支化聚乙烯亚胺分子链上存在伯、仲、叔氨基三种基团,使其呈碱性和阳离子活性,更容易与亲电试剂反应或者与氨基化合物反应改性。本申请的制备方法无需真空反应环境或惰性气体氛围,仅在无溶剂、无催化剂的熔融环境中进行开环聚合反应,合成条件温和,反应无环境污染,无副产物,n-磺酰基氮丙啶的转化率高(>99%),改性聚乙烯亚胺聚合物的收率高(>99%),纯度高(>98%),无需分离纯化,有利于大规模的工业生产。
优选的,所述n-磺酰基氮丙啶化合物具有式(ⅲ)所示结构;
其中,r为h、碳原子数为1~15的烷基、r2为甲苯磺酰基、甲磺酰基或吡啶磺酰基。
优选的,所述支化聚乙烯亚胺具有式(ⅳ)所示结构;
其中,m、n为正整数,10≤m+n≤600。
优选的,所述支化聚乙烯亚胺的重均分子量为600~25000。
优选的,所述支化聚乙烯亚胺和所述n-磺酰基氮丙啶化合物的摩尔比为1:(0.1~10)。
本申请中,在摩尔比为1:(0.1~1)时,熔融反应制得双亲性改性聚合物;在继续增大n-磺酰基氮丙啶化合物的用量时,熔融反应制得油溶性改性聚合物。
优选的,所述熔融反应的温度为120~200℃,时间为30~90min。
综上所述,本申请提供了一种改性聚乙烯亚胺聚合物及其制备方法。改性聚乙烯亚胺聚合物包括作为主链的支化聚乙烯亚胺以及接枝在支化聚乙烯亚胺上的磺酰仲胺或聚磺酰胺,为浅黄色非粘稠固体,相较于商业化的聚乙烯亚胺更容易实现后加工处理。通过调整支化聚乙烯亚胺和n-磺酰基氮丙啶的反应摩尔比例,可选择性制备双亲性和油溶性的改性聚乙烯亚胺聚合物。油溶性聚合物表现出优异的荧光性能,双亲性聚合物用于金属离子吸附的吸附效率高,且回收再利用时吸附效率基本不变,在吸附材料、分析检测等领域具有较大的应用前景。本申请的制备方法无需真空反应环境或惰性气体氛围,仅在无溶剂、无催化剂的熔融环境中进行开环反应或开环聚合反应,合成条件温和,无需透析、干燥等分离纯化操作,收率高,产物纯度高,有利于大规模的工业生产。
附图说明
为了更清楚地说明本申请实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。
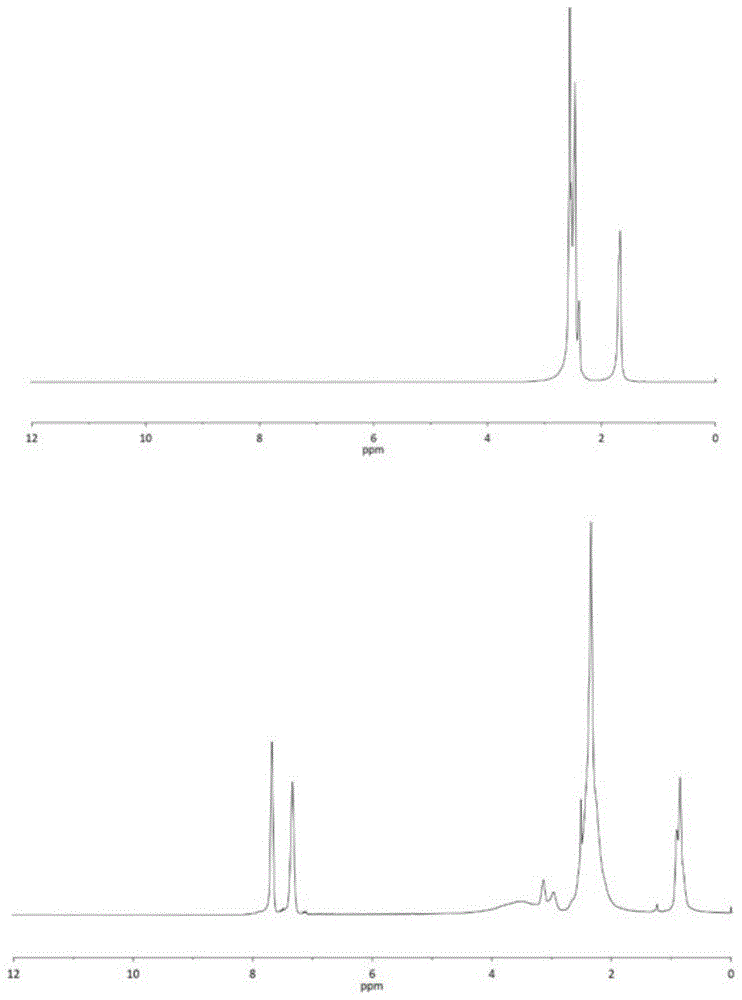
图1为本申请实施例4中所使用聚乙烯亚胺(上)以及制得产物(下)的核磁共振氢谱图;
图2为本申请实施例5中所使用聚乙烯亚胺(上)以及制得产物(下)的核磁共振氢谱图;
图3为本申请实施例4中所使用聚乙烯亚胺(a)、制得产物(b)以及实施例7中制得产物(c)的红外光谱图;
图4为本申请实施例5中制得产物的凝胶色谱图;
图5为本申请实施例2和实施例7制得产物以及分子量为1800的支化聚乙烯亚胺的荧光光谱图。
具体实施方式
为使得本申请的目的、特征、优点能够更加的明显和易懂,对本申请实施例中的技术方案进行清楚、完整地描述,显然,下面所描述的实施例仅仅是本申请一部分实施例,而非全部的实施例。基于本申请中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本申请保护的范围。
本申请实施例中所使用的支化聚乙烯亚胺为市售,本申请实施例中的n-磺酰基氮丙啶由相应的氮丙啶与磺酰氯试剂反应制备。本申请实施例中支化聚乙烯亚胺和氮丙啶化合物的摩尔比相对于聚乙烯亚胺的重复单元计算,氮丙啶化合物的r为甲基,r2为对甲苯磺酰基。
实施例1
称取摩尔质量比为1:0.3的支化聚乙烯亚胺(分子量为1800)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应0.5h,得到改性聚乙烯亚胺聚合物。制得的产物通过核磁共振进行结构表征为n-磺酰基氮丙啶亲核开环后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,且无需繁琐的透析、干燥等纯化操作。
实施例2
称取摩尔质量比为1:0.5的支化聚乙烯亚胺(分子量为1800)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应0.5h,得到改性聚乙烯亚胺聚合物。制得的产物通过核磁共振进行结构表征为n-磺酰基氮丙啶亲核开环后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例3
称取摩尔质量比为1:0.3的支化聚乙烯亚胺(分子量为10000)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应0.5h,得到改性聚乙烯亚胺聚合物。制得的产物通过核磁共振进行结构表征为n-磺酰基氮丙啶亲核开环后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例4
称取摩尔质量比为1:0.5的支化聚乙烯亚胺(分子量为10000)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应0.5h,得到改性聚乙烯亚胺聚合物。本申请实施例中所使用聚乙烯亚胺(上)以及制得产物(下)的核磁共振氢谱图如图1所示,所使用聚乙烯亚胺(a)和制得产物(b)的红外光谱图如图3所示,表明制得的产物为n-磺酰基丙啶亲核开环后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例5
称取摩尔质量比为1:1的支化聚乙烯亚胺(分子量为10000)和n-磺酰基基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应0.5h,得到改性聚乙烯亚胺聚合物。本申请实施例中所使用聚乙烯亚胺(上)以及制得产物(下)的核磁共振氢谱图如图2所示,制得产物的凝胶色谱图如图4所示,表明制得的产物为n-磺酰基氮丙啶亲核开环后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例6
称取摩尔质量比为1:5的支化聚乙烯亚胺(分子量为600)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应1.5h,得到改性聚乙烯亚胺聚合物。制得的产物通过核磁共振进行结构表征为n-磺酰基丙啶阴离子开环聚合后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例7
称取摩尔质量比为1:10的支化聚乙烯亚胺(分子量为600)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应1.5h,得到改性聚乙烯亚胺聚合物。本申请实施例中制得产物(c)的红外光谱图如图3所示,表明制得的产物为n-磺酰基氮丙啶阴离子开环聚合后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例8
称取摩尔质量比为1:5的支化聚乙烯亚胺(分子量为1800)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应1.5h,得到改性聚乙烯亚胺聚合物。制得的产物通过核磁共振进行结构表征为n-磺酰基氮丙啶阴离子开环聚合后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
实施例9
称取摩尔质量比为1:7的支化聚乙烯亚胺(分子量为1800)和n-磺酰基氮丙啶化合物加入10ml的试剂瓶中,等油浴锅或加热板或加热套温度到达200℃后,将试剂瓶放入其中搅拌30秒使n-磺酰基氮丙啶化合物和支化聚乙烯亚胺混合均匀,维持200℃在常压下静置反应1.5h,得到改性聚乙烯亚胺聚合物。制得的产物通过核磁共振进行结构表征为n-磺酰基氮丙啶阴离子开环聚合后的聚乙烯亚胺改性共聚物,转化率达到100%,纯度>98%,无需繁琐的透析、干燥等纯化操作。
对比实施例1~5以及实施例6~9制得产物的表征图谱,表明n-磺酰基氮丙啶发生亲核开环或者阴离子开环聚合取决于其与支化聚乙烯亚胺中的氨基比例。当支化聚乙烯亚胺的氨基含量远大于n-磺酰基氮丙啶(实施例1~4)时,n-磺酰基氮丙啶会与支化聚乙烯亚胺的伯胺和仲胺发生亲核开环反应,即在每摩尔当量的氨基上接枝一个氮丙啶组分;当聚乙烯亚胺氨基比例少于磺酰基氮丙啶(实施例6~9)时,叔胺作为有机碱会继续催化n-磺酰基氮丙啶发生阴离子开环聚合反应,即在每摩尔当量的氨基上接枝由多个氮丙啶聚合后的组分。结果表明,仅需通过调整支化聚乙烯亚胺和n-磺酰基氮丙啶的反应摩尔比例,便可得到接枝链量以及接枝长度不同的双亲性或油溶性的改性聚乙烯亚胺聚合物。
测试例1
取实施例2和实施例7制得的产物,以及分子量为1800的支化聚乙烯亚胺。各称取等摩尔质量的上述原料溶解在四氢呋喃和水比例为4:1的混合溶液中,再通过荧光光谱仪测试荧光性能,本申请实施例2和实施例7制得产物以及数均分子量为1800的支化聚乙烯亚胺的荧光光谱图如图5所示。
图5表明,实施例2制得产物的荧光强度与未改性支化聚乙烯亚胺的区别不大,但在458nm左右的荧光强度稍微变强,说明n-磺酰基氮丙啶开环接枝到支化聚乙烯亚胺引起其在458nm荧光强度的增强。而实施例7相比于未改性支化聚乙烯亚胺的荧光强度在458nm上增强了10倍,说明引发n-磺酰基氮丙啶发生阴离子开环聚合反应后能够明显提升接枝量,而接枝链的增加能提高支化聚乙烯亚胺的荧光强度。
测试例2
配置含有浓度均为1克每升的铁、铜、锌、铬、镍、钴和锰的离子溶液,各取实施例1~9制得的产物30毫克,分别加入至配置好的1毫升的离子溶液中搅拌3小时,离心去除固体后稀释上澄清液,通过原子吸收光谱仪测试得到各金属吸附前后的离子浓度,实施例1~9制得产物吸附前后离子溶液中金属离子浓度的变化情况如表1所示。
表1实施例1~9制得产物吸附前后离子溶液中金属离子浓度的变化情况
表1表明,实施例1-4制得的产物对各金属离子的吸附效率达到了100%,具有很强的吸附性能,且不存在分离回收困难、易流失等缺点。需要说明的是,实施例1~4中由于接枝量过少,材料偏向亲水性可能导致在金属吸附过程中一少部分材料溶于水造成少许实验误差,导致测得的吸附效率均为100%。实施例6-9制得的产物的吸附效率较低,可能由于聚乙烯亚胺占比较少而导致吸附性能下降。实施例5制得的产物的金属吸附效率对铜、铁和锌较好,具有吸附选择性。结果表明,相比较于未改性的水溶性聚乙烯亚胺,改性后的聚乙烯亚胺聚合物呈固态非粘稠液体,不溶于水,更容易操作和分离回收,且不易流失,能够用于金属离子吸附,且吸附效率高。
测试例3
取30毫克实施例5制得的产物,加入到1毫升浓度为1克每升的铁离子溶液中搅拌3小时,离心分离上澄清液,固体用1%的稀硝酸和去离子水清洗,清洗后继续加入1毫升铁离子溶液搅拌3小时,上述步骤重复5次,每次离心后的上澄清液稀释后用原子吸收光谱仪测试金属离子浓度,循环5次实施例5制得产物对铁离子的吸附效率如表2所示。
表2循环5次实施例5制得产物对铁离子的吸附效率
表2表明,实施例5制得产物连续吸附5次的吸附效率基本不变,循环使用后的吸附效率稍微下降可能由于分离上澄清液和清洗固体时固体损失和部分稀硝酸未完全清除导致。说明改性后的聚乙烯亚胺聚合物能回收利用,且性能基本保持。
以上所述,以上实施例仅用以说明本申请的技术方案,而非对其限制;尽管参照前述实施例对本申请进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本申请各实施例技术方案的精神和范围。




